Amber MD关键字
之前了解了一遍Amber动力学输入文件中关键字的含义,但是几个月不用再一看又不熟悉了。为此做一个方便快速检索参考的中文笔记。
MD模拟
输入关键字
这是我使用的production步的输入文件
Equilibration:constant pressure |
其中功能相关的设置被放置在相近的位置
- imin
选择任务类型1(最小化)2(MD)5(读取轨线进行再次计算) - ntx
坐标(-c指定)读取方式1(仅读取笛卡尔坐标)5(读取坐标和速度以进行重启) - irest
读取rst文件(-c指定)的行为0(不重启模拟,不读取速度,时间设置为0,开始新的模拟)1(重启模拟,从rst文件中读取坐标和速度,时间继续,不开始新的模拟)
- ntpr
out和mdinfo文件的写入频率
每ntpr步写入一次,默认为50 - ntwx
轨线文件的写入频率
每ntwx步写入一次,默认是0(不写入) - ntwr
rst文件的更新频率,减少任务崩溃的损失
每ntwr步更新一次,任务结束后也更新一次,默认值等于nstlim(只在任务结束写入一次) - iwrap
写入到轨线和rst文件中时分子的绝对位置和相对位置:1(写入每个分子在初始盒子中对应的等价分子,利用平移对称性只用初始盒子的大小描述体系)0(写入每个分子的绝对位置,往往用于扩散分析,在1的情况下无法分析扩散)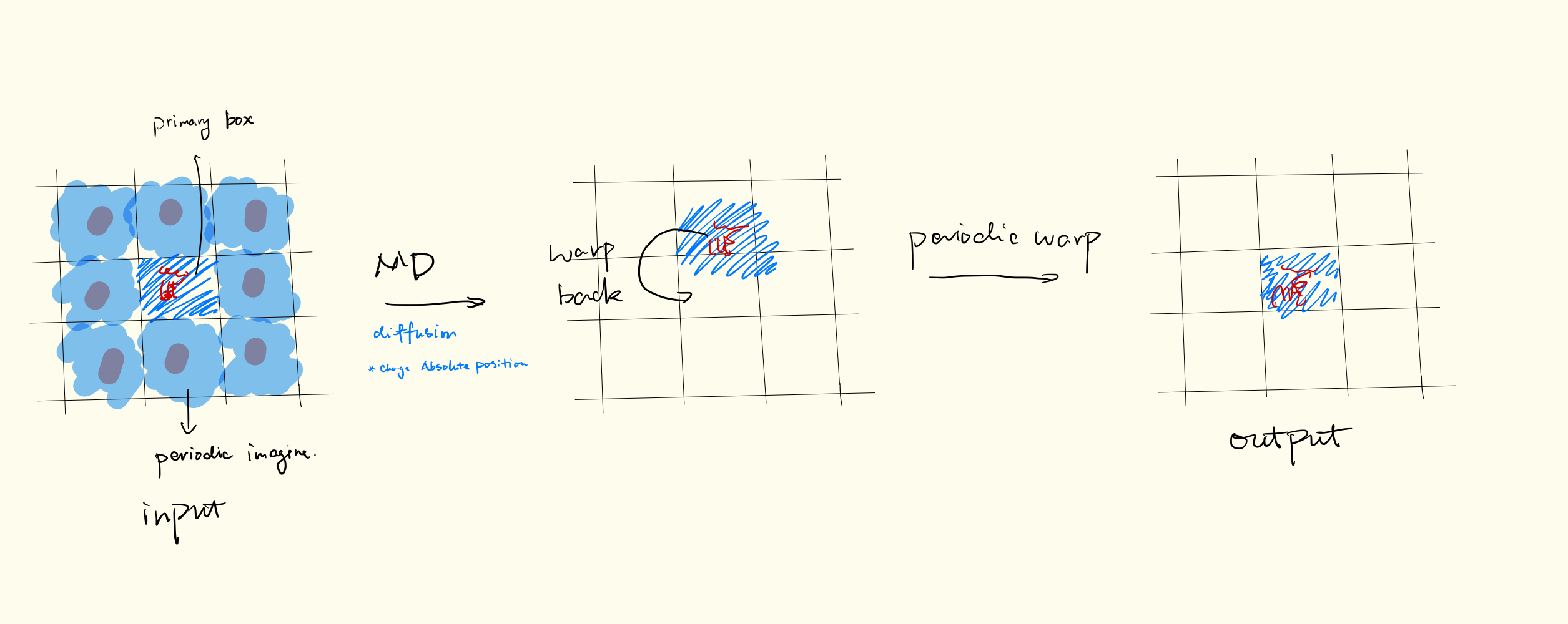
类似贪吃蛇
*不涉及扩散分析的任务都建议使用iwarp=1
ntf与ntc
SHAKE键振动限制,往往设置NTF=NTC
目的:
MD的基础步长受体系中最快的运动限制,限制最快的X-H振动以获取更大的步长
TIP3P也是在限制X-H振动的情况下拟合的,使用该力场需要加对应限制1(不使用SHAKE)2(所有含H的键被限制,在dt=0.002时必用)3(所有键长都被固定)nstlim
MD的步数dt
MD的步长,单位:ps,默认为0.001,加氢SHAKE往往使用0.002cut
描述nonbonded截断的范围 单位:埃
范围内静电作用使用精确的 QQ/r^2 范围外用Particle Mesh Ewald (PME)方法近似
有文献指出需要大于等于10.0,尤其是计算蛋白相互作用时
ntt
温度控制0(体系能量固定,绝热,NVE系综)1(恒定温度,使用weak-coupling算法:只保证总动能不变,将超出的动能重新分配。不能保证温度在分子各处一致,往往导致低频自由度能量过高,无法保证各自由度间的能量关系)2(恒定温度,使用Anderson-like算法:每隔vrand步就模拟碰撞,随机的重置体系中的速度以符合设定温度。会导致轨线没有绝对时间相关的意义,只有在vrand步之内可以进行绝对时间相关的分析,vrand值太小会导致构象空间的探索变慢(默认1000))3(恒定温度,使用Langevin算法:使用gamma_ln设置的$\gamma$值,有时会崩溃?)9(恒定温度,使用优化的Isokinetic Nose-Hoover chain ensemble (OIN)方法:貌似是为了使用一些加速步进的方法?)10(恒定温度,使用Stochastic Isokinetic Nose-Hoover RESPA integrator方法:貌似是为了使用一些加速步进的方法?Tuckerman组开发)11(Berendsen控温器,之后用到了再补充)nmropt
0(默认)无nmr分析1读入NMR限制和权重变化 (控制恒温器温度变化)2读入NMR限制、权重变化、NOESY volumes、化学位移、残基偶极限制temp0
设定恒定温度值,大于300时需要降低步长tempi
初始温度,用于heat步,初始速度使用maxwellian分布
可增加温度控制段,设置在不同阶段温度的变化,如:heat.in >folded &wt
type = 'TEMP0',
istep1= 0, istep2=18000,
value1= 0.0, value2=300.0, #在前18000步升温至300K
/
&wt
type = 'TEMP0',
istep1= 18001, istep2=20000,
value1= 300.0, value2=300.0, #在后2000步保持恒温
/
&wt
type = 'END',
/
ig
决定初始速度的随机种子,也决定了Langevin和Anderson-like方法中的伪随机值-1(时间相关的随机种子)默认其他值(固定种子,一般只在调试时使用)
ntp
压力控制,逐渐调整cell体积以达到设定压力0(无压力控制,默认)1(使用各向同性位置修正)2(使用各向异性位置修正,只能用于正交盒子,一般用于膜系统的模拟)3(使用半各向异性位置修正,只能用于正交盒子和恒定表面张力体系,将表面切向的两个压力耦合在一起)pres0
恒定压力值,单位bar,默认为 1.0ntb
周期性边界的类型, Amber20中不需要单独设置0(无周期性边界 关闭PME 微观系综)1(定容)2(定压)ntr
用简谐势在笛卡尔空间中限制原子位置
需要指定参考结构-ref目标原子 和 力常数0(不限制)1(限制)restraint_wt
位置限制的权重,单位:$kcal·mol^{−1}·Å^{−2}$
设置$k(\Delta x)^2$ 中的k值restraintmask
描述目标原子,
语法:- 描述基元
起始符:起始选择残基@起始描述原子
序号/名称选择
可以用,分隔和-连接的序号描述 例::1-10,12和@12,54-85
可以用残基/原子名称描述 例::ARG,ALA和@CA,C,O
可以用原子类型描述 但是需要在@后加%例:@%N*,N3
可以用:*描述所有结构,注意!不是通配符,貌似是由于有些原子类型里包含星号如C*
可以用=充当通配符,但是只能在字符之后 例:@H=描述所有H原子 - 描述基元间
逻辑关系&或|与!非
距离关系<:数字与>:数字在数字范围之内/之外的所有残基<@数字与>@数字在数字范围之内/之外的所有原子
*可以用()描述结合的优先级
可以用ambmask测试
ambmask -p prmtop -c inpcrd -out [short| pdb| amber] -find [maskstr]
会输出选中原子到stdout- 描述基元
施加基于内坐标的constraint
NMR restraint的方案
- 在.in文件中加入
nmropt=1关键字,在结尾添加DISANG={path-to-rs-file} - 制作.rs文件描述restraint
&rst
iat={atom_idx},{atom_idx}, r1=抛物线势左起点, r2=抛物线势左终点, r3=抛物线势右起点, r4=抛物线势右终点, rk2=左边抛物线力常数, rk3=右边抛物线力常数,
ir6={指定一组原子时确定中心的方式}, ialtd={在势极大时的行为/是否将偏离过大的结构拖回}
&end
验证MD结果是否有效
查看out文件中的参数随轨线步数变化
使用process_mdout.perl脚本处理
perl process_mdout.pl 文件名 |
用cpptraj查看RMSD
parm *prmtop |
如果在prod步之前密度、温度、能量、rmsd都收敛,同时prod中这几项稳定
则认为prod产生的数据可以用于采样
关于提升GPU下的计算效率
Amber文档中给出的最大化GPU加速效率的建议:
- 避免使用过高的写入频率(小的NTPR, NTWX, NTWV, NTWE, NTWR)
- 避免$ntave \neq 0$
- 避免在不必要时使用NPT系综,考虑使用Monte Carlo barostat (barostat=2)
- 使用gbsa=3而不是gbsa=1,前者适合GPU
- 使用langevin温度控制会稍微降低速度
MMPBSA 与 MMGBSA
将结合自由能拆分成气相的结合自由能和三个物种各自的溶剂化自由能
$\Delta G_{solv,bind}^0 = \Delta G_{bind,vacuum}^0+\Delta G_{solv,complex}^0−(\Delta G_{solv,ligand}^0+\Delta G_{solv,receptor}^0)$
GB(Generalized Born)和PB(Poisson-Boltzman)描述不同的求解溶剂化自由能中静电部分的方法
使用LCPO方法求解疏水部分的贡献
J. Comput. Chem., 2016, 37, 2436–2446
关于参数选取
准备文件
需要准备三部分文件:
轨线(.nc)
描述配体、受体、复合物和溶剂化复合物的拓扑文件(.prmtop)
为PB/GB设置合适的
PBRadii
该设置(只作用用隐式溶剂模型)不影响常规(即使用显式溶剂)MD计算 相关讨论
可以通过set修改leap的全局设置之后生成新的prmtop文件set default PBRadii mbondi2
也可以通过ParmEd修改现有的prmtop文件
parmed -p <your_prmtop> -i input_file
input_file中为
parmed.in changeRadii mbondi2
parmout new_radii.prmtop参数参考
值 igb值 bondi 7 mbondi 1 mbondi2 2 or 5 mbondi3 8 分别为三部分结构生成不含溶剂的拓扑文件
同时需要了解体系的离子强度
设置输入文件(.in)
格式类似MD的输入文件$AMBERHOME/bin/MMPBSA.py --input-file-help可以查询可设置选项MMPBSA.in Sample input file for GB and PB calculation
&general
startframe=5, endframe=100, interval=5,
verbose=2, keep_files=0,
/
&gb
igb=5, saltcon=实际离子浓度,
/
&pb
istrng=实际离子浓度, fillratio=4.0,
/startframe&endframe描述开始和结束的帧interval步长keep_files默认是1(保留所有轨迹文件和mdout)0(删除所有)2(保留所有)
(*关于巨大的reference.frc 重编译有些时候不太显示,希望之后amber能给个选项)verbose0(只输出差值部分)1(也输出组分的数据)2(增加bonded terms)
saltcon 盐浓度(单位:M)
istrng 离子强度(单位:M)
fillratio 矩形有限差分网格的最长维度与溶质的最长维度之比。对于与水尺度类似的溶剂一般不用改
gb和pb部分设置对应的参数
运行
$AMBERHOME/bin/MMPBSA.py -O -i MMPBSA.in -o FINAL_RESULTS_MMPBSA.dat -sp complex_solvated.prmtop -cp complex.prmtop -rp receptor.prmtop -lp ligand.prmtop -y *.mdcrd |
或者使用并行版
mpirun -np 核数 $AMBERHOME/bin/MMPBSA.py.MPI -O -i MMPBSA.in -o FINAL_RESULTS_MMPBSA.dat -sp complex_solvated.prmtop -cp complex.prmtop -rp receptor.prmtop -lp ligand.prmtop -y *.mdcrd |
TIPS for COMET USER
在comet上没有直接的MMPBSA.py.MPI文件 经过咨询了解到可以直接讲MMPBSA.py复制到自己目录下为MMPBSA.py.MPI
参考的提交脚本:RunMMPBSA.sh >folded
> #SBATCH --job-name=name
> #SBATCH --partition=compute
> #SBATCH -N 1
> #SBATCH --ntasks-per-node=24
> #SBATCH --mem=100GB
> #SBATCH -t 48:00:00
> #SBATCH --signal=B:SIGUSR1@60
> #SBATCH --verbose
> #SBATCH --hint=nomultithread
> #SBATCH --mail-user=mail
> #SBATCH --mail-type=all
>
> #Run MMPBSA
> module purge
> module load amber
> module load intel
> module load mpi4py
>
> ibrun python3 /path/to/MMPBSA.py.MPI .... options ...
>
计算结果
计算会在当前目录下输出
| *.mdcrd | GB和PB计算过程中分析过的坐标 |
| *.mdout | 所有指定帧的能量 –> 并行按后缀依次合并即可 |
| *.pdb | 平均结构的PDB文件,可进行准简谐熵计算 |
| FINAL_RESULTS_MMPBSA.dat | 结果输出文件 |
其中结果输出文件中会输出组分能量项:
VDWAALS |
$H_{MM,vdw}$分子力场能的vdw部分 |
EEL |
$H_{MM,ele}$分子力场能的静电部分 (不包含周期性边界条件cut=999.0) |
EPB/EGB |
$E_{solv,ele}$溶剂化自由能的静电部分,由PB或GB计算 |
ESURF |
$E_{solv,nonpolar}$ ??有什么区别 (GB only) |
ENPOLAR |
$E_{solv,nonpolar}$ 非极化作用的空穴部分 // 在mdout文件里也被写做ECAVITY (PB only) |
EDISPER |
$E_{solv,nonpolar,disper}$ 非极化作用的色散部分 (PB only) |
其中并行相关的帧分配方式:将所有帧连续的分成MPI.COMM_WORLD.get_size()块之后,块的大小通过平均后渐进的方式决定
# Sanity check |
- 根据mdout文件的逐帧分析/作图脚本
https://github.com/shaoqx/CalcKit/tree/master/Misc_scripts/MMPBSA
提取每一帧各能量项的数值,可以容易的分析和作图
MMPBSA中包含的熵
分子力场项 - 不包含熵
溶剂静电PB项 - 不包含熵
溶剂非极性部分(inp=2) - 1. 形成空穴(溶剂内聚能破坏,溶剂熵减)2. 溶质溶剂吸引(VDW作用)
MMPBSA中的周期性边界条件
查看具体的模块发现计算MM部分能量时使用的是无周期性边界条件(igb>0时默认关闭周期性边界条件ntb=0,cut=999.0)
这就意味着盒子的大小和形状会对结果产生影响(离子的位置?)
!!如果用长方形盒子必然会导致GPU在equi步报错!!
关于各种选项
根据Expert Opin Drug Discov. 2015 May 4; 10(5): 449–461.一文,总结一些在用MMPBSA研究新体系的思考:
- 一般可选择的有:
- PB/GB
- 3A/1A
- 关键水的处理
- 计算熵的方法
根据文中的建议,这些选择和对研究的具体体系有一定的依赖性,最好在研究之前benchmark
- 采样方法:
文中建议使用多次短模拟合并采样的方法,以降低SD为标准,正在测试其可靠性
使用循环多次跑prod步之后用cpptraj合并for i in {1..20}; do pmemd.cuda -O -i ./MD/prod.in -o ./MD/prod${i}.out -p *.prmtop -c ./MD/equi.rst -ref ./MD/equi.rst -r ./MD/prod_${i}.rst -x ./MD/prod_${i}.nc; done
对比;parm ../*.prmtop
trajin prod*.nc 200 last
autoimage
trajout prod.nc
go- 跑100ns取后90ns,每10ps一帧取9000帧分析 (结果比较接近实验值)
- 跑100ns取后90ns,每30ps一帧取3000帧分析 (结果比较接近实验值)
- 跑20条5ns 每个轨迹取后4.5ns,每10ps一帧合起来共9000帧 (结果很差)
- 跑20条5ns 每个轨迹取后4.5ns,每30ps一帧合起来共3000帧 (结果很差)
非标准残基的参数化
从PDB中复制非天然氨基酸的几行保存为新文件(每类一个即可) 注意需要把空缺的化合价补全,一般最后是一个独立氨基酸的结构,否则antechamber难以正确判断原子类型
使用antechamber生成ac文件
antechamber -fi pdb -i {输入路径.pdb} -fo ac -o {输出路径.ac} -c bcc -at amber -nc {净电荷数}
“Note that antechamber is mostly set up to use gaff atom types, so it occasionally makes a mistake with the Amber atom types.”
这要怎么自动化啊,问题看起来是随机的,要怎么确定什么原子类型是正确的?貌似每次都会把N给错
*注意生成的ac文件会修改原子名称,会将所有数字开头的原子名称修改制作描述和蛋白其他部分连接方式的mc文件(mianchain)
XXX.mc >folded HEAD_NAME {连接前一个氨基酸的原子名,如N1}
TAIL_NAME {连接后一个氨基酸的原子名,如C3}
MAIN_CHAIN {从head到tail依次的原子名}
MAIN_CHAIN {...}
MAIN_CHAIN {...}
MAIN_CHAIN {...}
OMIT_NAME {需要从最终结构中删除的原子,如某些构建方式中会多出来的H}
OMIT_NAME {...}
OMIT_NAME {...}
OMIT_NAME {...}
PRE_HEAD_TYPE {前一个氨基酸用来连接改氨基酸的原子类型,如C}
POST_TAIL_TYPE {后一个氨基酸用来连接改氨基酸的原子类型,如N}
CHARGE {净电荷数}使用prepgen生成prepin文件
prepgen -i {输入路径.ac} -o {输出路径.prepin} -m {mc文件路径.mc} -rn {残基名}
使用frcmod第一次使用天然氨基酸的参数(ff14SB)参数化
parmchk2 -i {输入路径.prepin} -f prepi -o {输出路径.frcmod} -a Y -p $AMBERHOME/dat/leap/parm/parm10.dat
将没有参数化的部分用gaff生成的参数补全
grep -v "ATTN" {路径.frcmod} > {路径.frcmod1} # Strip out ATTN lines
parmchk2 -i {路径.prepin} -f prepi -o {路径.frcmod2}最后得到两个参数文件
使用prepin和两个frcmod文件通过tleap完成最后的参数整合
MCPB.py的使用
参考官网教程
http://ambermd.org/tutorials/advanced/tutorial20/mcpbpy.htm
相关教程
http://bbs.keinsci.com/thread-26531-1-1.html
MCPB.py [flags] [options]
-i 输入文件
-s 步骤数
目标
参数化金属中心(MetalUnit)以及附近的配体残基(非标准质子态)(非标准连接方式)
流程
大致流程是:
- 制作目标蛋白模型
- 从目标模型中提取出各种参数化相关的组件(mol2格式)
- 使用MCPB.py根据提供的组件自动生成Gaussian输入文件
- Gaussian计算,利用MCPB.py自动读取输出文件中的信息完成参数化
I. 制作目标蛋白模型(preparation)
根据研究需求制作目标蛋白的MD-ready模型
要求:
- 无原子缺失(蛋白,cofactor,底物,水 的轻重原子)
- 正确的质子态(确定配体、水的质子朝向合理)
- cofactor处于合理的binding位置
- 底物处于合理的binding位置
- 包含必要的active site water
转换为amber格式的pdb并重置index
pdb4amber -i {输入路径} -o {输出路径} |
II. 提取目标蛋白模型组件(mol2文件)
以及非天然氨基酸和配体的frcmod参数文件
“MCPB.py uses mol2 files based on the residue names” - 只要保证各组件残基名称和PDB中保持一致即可
“You need to create different mol2 files for different TYPES of metals, water, and NCAA in the PDB file” - 每类组件只需要一个mol2文件
金属离子
- 从蛋白的PDB中,复制金属离子的一行保存为新文件(一个原子即可)
- 运行metalpdb2mol2.py脚本,指定电荷,并生成mol2文件
python metalpdb2mol2.py -i {输入路径} -o {输出路径} -c {电荷数}
Active site water 水
- 从PDB中复制水的几行保存为新文件(一个水即可)(*需要氢和朝向正确)
- 使用antechamber生成mol2文件
antechamber -fi pdb -fo mol2 -i WAT_H.pdb -o WAT.mol2 -at amber -c bcc -pf y
- 将mol2中的原子类型从oh和ho修改为OW和HW (例)
配体(cofactor和底物)
- 从PDB中复制配体的几行保存为新文件(每类一个配体即可)
- 使用antechamber生成mol2文件*注意最后保存的mol2文件的名称要和PDB中配体的残基名一致
antechamber -fi pdb -fo mol2 -i {输入路径} -o {输出路径} -c bcc -pf y -nc {配体净电荷数}
- 生成frcmod文件
parmchk2 -i {输入路径} -o {输出路径} -f mol2
非天然氨基酸(modified AA)
参考”非标准残基的参数化”一节
生成prepin,frcmod1,frcmod2文件将prepin文件转换为mol2文件
antechamber -fi prepi -i {输入路径.prepin} -fo mol2 -o {输出路径.mol2} -nc {净电荷数}
*注意最后保存的mol2文件的名称要和PDB中配体的残基名一致
*检查mol2中的原子类型名称和prepin中的是否一致
III. 使用MCPB.py生成Gaussian输入文件
配置MCPB计算
制作.in配置文件mcpb.in >folded original_pdb {完整模型路径.pdb}
group_name {自定义一个名字}
cut_off {参数化区域选择范围,默认填2.8}
ion_ids {金属的原子序号 (空格分隔可以多个,但是只能是一个金属complex里的多个)}
ion_mol2files {金属的mol2文件 (空格分隔可以多个,但是只能是一个金属complex里的多个)}
naa_mol2files {非天然氨基酸的mol2文件 *配体、水、cofactor、modified AA (空格分隔可以多个)}
frcmod_files {补充的参数文件 *配体、cofactor、modified AA (空格分隔可以多个)}
large_opt {是否优化大模型,默认填1 (1:只优化H、0:不优化)}
software_version {g16或g09}不在金属中心附近的mol2和frcmod文件影响什么 我不小心没加上但是也没有报错?
- 不在目标金属中心附近的NCAA的mol2和frcmod文件貌似不影响结果,测试了在mcpb.in中加和不加两种情况得到的最终的mol2文件和frcmod文件没有区别
貌似只影响最后生成的tleap.in文件
- 不在目标金属中心附近的NCAA的mol2和frcmod文件貌似不影响结果,测试了在mcpb.in中加和不加两种情况得到的最终的mol2文件和frcmod文件没有区别
运行MCPB生成com文件
MCPB.py -i {路径.in} -s 1
此步会生成小模型、标准模型和大模型的PDB和fingerprint文件。还会生成小模型和大模型的高斯输入文件。
- 大模型 的高斯输入文件将首先对氢原子进行优化,以纠正任何放置不当的氢原子。
- 标准模型fingerprint文件(1mlw_standard.fingerprint)包含标准模型中原子的原子类型信息(第3列是原始原子类型,最后一列是最终分配的原子类型)。金属离子的原子类型(名称以 M 开头)和连接原子(名称以 Y/Z 开头)会自动重命名,以将它们与 AMBER 力场中的其他原子类型区分开来。(为了参数化后的识别)
- 可以将 step_number 设为 1n 来强制不重命名任何原子类型并生成fingerprint文件,然后手动修改(在此之前,请检查 AMBER force field parm*.dat 文件以确保金属离子和连接原子类型 不与amber已有的原子类型冲突)。
- fingerprint文件在文件末尾还包含金属离子与周围原子之间的链接信息,开头字母为“LINK”。用户还可以根据自己的需要手动修改它们(例如,如果 QM 几何优化后有任何连接更改)
- 运行Gaussian计算
MCPB.py生成的输入文件默认用的是B3LYP/6-31G*、2核、3000MB。建议至少改为B3LYP-D3并根据实际资源修改核数和内存设置
注意!默认的自旋多重度可能有误,需要检查并修正!
g16 < 1OKL_small_opt.com > 1OKL_small_opt.log |
这步得到的3个log文件和fchk文件需要被放置在与第一次运行MCPB.py相同的路径
*需要检查优化得到的结构中配位键是否断裂,如果断裂需尝试:
- 更换理论层级
- 先固定优化其他坐标之后放开优化配位键
IV. 使用MCPB.py读取Gaussian输出文件并生成参数
生成frcmod参数文件
在运行第一步的目录运行MCPB.py -i 1OKL.in -s 2
*注意运行该步的时候目录下不能存在temp文件夹,该脚本貌似会生成一个temp文件
拟合电荷并生成mol2文件
MCPB.py -i 1OKL.in -s 3
默认使用charge model B
生成tleap输入文件和修改残基名后的PDB
MCPB.py -i 1OKL.in -s 4
获得
- {名称}_mcpbpy.pdb
- {名称}_tleap.in
V. 运行tleap.in完成参数化
tleap -s -f {名称}_tleap.in |
需要在tleap.in文件中加入所有非天然氨基酸、配体的参数对应命令
可以改为用protein.ff14SB和water.TIP3P吗?
为什么会报错Residues lacking connect0/connect1? 貌似是使用mol2和bond组合的正常表现 这个问题会导致直接用tleap生成的PDB存在错误的TER标记,但是参数经过parmed检查是正确的
V-1. (对于多聚体)修改pdb和tleap以适配其他金属中心
对于多聚体,其他对称的金属中心可以使用其中一个金属中心MCPB得到的参数
但是需要修改tleap.in和PDB使tleap可以讲其识别到对应的参数
修改_mcpbpy.pdb中其他金属中心的残基名
参考MCPB.py第四步中输出的改名位点,例:XXX.mc >folded Renamed residues includes: 519-HID
Renamed residues includes: 521-HID
Renamed residues includes: 684-HIE
Renamed residues includes: 1441-CU
Renamed residues includes: 1445-WAT修改其他链中对应的名称
修改_tleap.in中的bond语句
为其他链的金属中心和配体添加对应的键,例:XXX.mc >folded bond mol.519.NE2 mol.1441.CU
bond mol.521.NE2 mol.1441.CU
bond mol.684.ND1 mol.1441.CU
bond mol.1441.CU mol.1445.O
bond mol.518.C mol.519.N
bond mol.519.C mol.520.N
bond mol.520.C mol.521.N
bond mol.521.C mol.522.N
bond mol.683.C mol.684.N
bond mol.684.C mol.685.N
bond mol.1239.NE2 mol.1446.CU
bond mol.1241.NE2 mol.1446.CU
bond mol.1404.ND1 mol.1446.CU
bond mol.1446.CU mol.1450.O
bond mol.1238.C mol.1239.N
bond mol.1239.C mol.1240.N
bond mol.1240.C mol.1241.N
bond mol.1241.C mol.1242.N
bond mol.1403.C mol.1404.N
bond mol.1404.C mol.1405.N
V-2. (对于多个不同金属中心)修改pdb和tleap.in
MCPB.py新加的原子类型名称都使用默认名称导致重复,需要更改重复的原子类型名称以适配多次MCBP生成的mol2和frcmod文件
- 将重复的M1,Y1等原子类型重命名之后合并到tleap.in中
- 在frcmod, mol2文件中查找替换成修改后的原子类型名称
将mol2文件和frcmod文件合并至tleap.in中
在此步注意如果有重复的残基名(例如有两个不同的Ca2+中心时都会被命名为CA1)需要在mol2文件和tleap.in都更改为彼此独特的残基名(如CA2)
**注意 CA2 = loadmol2 xxxx.mol中等号左边的名字符合和pdb配对 一定要修改将bond指令合并至tleap.in中
将更新后的残基名合并到同一个pdb文件中
如果两个不同的金属中心彼此比较接近/有共享的配体
则难以以此方式合并参数,会引起一系列的参数确实错误
有些时候对于辅助折叠的金属中心可以使用nonbonded model来描述
Misc
注意antechameber生成的mol2文件中的原子类型可能不规范 pymol可能会不识别从而转存的时候将原子名称搞错
- 教程中提到需要修复原子序号的问题,但是没有修复也不怎么影响模拟,跑了100ns都没问题
轨线输出文件格式
- mdcrd: ASCII编码的纯坐标 每帧由中心的三个坐标组成的一行分割 帧内由连续的每个原子的xyz坐标组成(每行10个数值)
amber.mdcrd >folded Cpptraj Generated trajectory
17.357 12.865 14.673 17.869 12.805 13.804 16.361 12.909 14.514 17.650
12.025 15.151 17.683 14.091 15.410 17.493 14.988 14.820 17.057 14.204
...
33.227 24.489 1.071 33.789 24.515 1.846 32.350 24.321 1.417
58.637 58.637 58.637
...
[next frame] - NetCrd(.nc/.crd): 二进制的坐标文件
可以通过cpptraj将这些格式以及pdb相互转换
Umbrella Sampling模拟与分析
目标
沿某个预定义的“路径坐标”采样从而得到体系沿该路径变化对应的自由能变化(有平均力势得出;Potential Mean Force)
路径坐标的定义由研究的问题决定并定义了得到的数据的意义
原理
将“路径坐标”划分为多个窗口,用势函数约束该窗口的坐标值进行采样(例如:简谐势)
如果在采样后各窗口间采样得到的几何结构在“路径坐标”上有足够重叠,则可以使用WHAM方法去除“约束势函数”带来的bias从而得到自由能曲线
流程
1. 平衡初始结构
与一般的MD模拟相同,参数化并运行min heat equi等步骤
2. 定义路径坐标
根据研究的问题定义路径坐标
除明确的几何坐标如二面角之外也可以定义一些近似的路径例如用口袋氨基酸骨架质心与配体质心的距离角度等近似binding的路径坐标
3. 选择Umbrella Sampling的窗口
原则
窗口之间在路径坐标上必须重叠(这要求力常数不能太大,间隔不能太大)
窗口要尽可能少
使用简谐势的话 力常数(也就是约束强度)需要足够大来保证采样足够充分(如果路径的内禀势能非常大,例如断强键,力常数需要匹配地大从而保证每个窗口各不相同)
如何先验地得到合适的力常数?
4. 在每个窗口进行MD模拟
以带几何约束的模拟相同,见施加基于内坐标的constraint
每个窗口进行 min,equi,prod 模拟
可以在.in文件中增加
&wt |
与
DUMPAVE=dihedral_60.dat |
部分从而直接输出采样得到的constraint的值
貌似不进行heating了,不过heating本身的必要性也不明确 (https://github.com/openmm/openmm/issues/4240)
运行时长
需要模拟足够长时间从而保证该约束条件下的采样收敛
理想情况下被约束的坐标自己往往涉及较大时间尺度的变化而其余的坐标可以在可接受的模拟时间尺度内收敛
*官方教程建议采样完成后随机去除20%的数据再计算一次PMF来验证其收敛
初始结构
初始结构在路径坐标上的值不宜与约束的目标值相差太大(例如:初始结构为1A 约束在10A)
但是也需要尽可能地平行提交计算任务
常用的策略是手动创建一些中间点的初始结构之后从这些结构开始
或者使顺序运行逐步改变初始结构
验证采样结果
可以做出采样结果在路径坐标上的histogram来判断设置是否有效
- 重叠是否充分
- 采样是否全面
5. 用WHAM分析结果
使用 Alan Grossfield 开发的WHAM程序
5.1 安装WHAM
从Alan Grossfield的官网上下载源代码(http://membrane.urmc.rochester.edu/?page_id=126/)
参考manual安装
tar -xvf wham-release-#####.tgz |
注意截至2.0.11版本,WHAM的编译仍不支持GCC 10.2.0,调整环境至GCC 8.2.0方编译成功
5.2 运行WHAM
{dir_of_wham}/wham [P|Ppi|Pval] hist_min hist_max num_bins tol temperature numpad metadatafile freefile [num_MC_trials randSeed] |
P|Ppi|Pval
指定反应坐标是周期性的(用于角度坐标)其中P指定360,Ppi指定2pi,Pval指定val的具体值(如P180.0)为一个周期hist_minhist_max
设置目标坐标的分布范围,所有范围外的点都被忽略num_bins
设置窗口数,对应最后自由能图上的点数tol
设置WHAM计算的收敛PMF阈值,单位为kcal/moltemperature
指定输入的采样数据对应的温度numpad
指定padding数据点的数量,对于周期性目标坐标起作用,该设置与非周期性目标坐标的计算无关metadatafile
指定输入的数据文件路径freefile
指定输出文件的路径num_MC_trialsrandSeed
如果指定,错误分析不进行,并使用Monte Carlop bootstrap 错误分析,分别指定fake data的数量和随机种子
用例
wham 14.3 35.7 84 0.01 300.0 0 meta.dat result.dat |
其中meta.dat的格式为
/path/to/timeseries/file loc_win_min spring [correl time] [temperature] |
其中
/path/to/timeseries/file
为数据文件路径loc_win_min
为该窗口的势函数的极小值点spring
为V = 0.5k(x-x0)^2定义下的力常数correl time
只在Monte Carlop bootstrap 错误分析时使用指定该采样集合的相关时间temperature
如果指定覆盖命令行指定的温度
5.3 分析WHAM结果
WHAM的结果为坐标和自由能,直接作图分析
Misc
tleap
tleap自动加氢甚至不会考虑周围的金属离子(如Zn2+等配位中心),会把旁边的残基质子化而不考虑配位
prepin格式
https://ambermd.org/doc/prep.html
利用树和LOOP的方式描述连接
GPU在equi步报错
用solvatebox的话就会用solvateOct则不会
迷惑的内存错误
Error: an illegal memory access was encountered launching kernel kClearForces |
参考
https://blog.csdn.net/Litedg/article/details/124230876
http://archive.ambermd.org/202203/0149.html 说了部分原理
主要的原因:
- GPU相关的MD精度有限,在涉及巨大的数值的时候会出现该问题
- 往往意味着结构中有bad contact从而存在巨大力
解决方案:
- 用CPU跑min步可以解决大部分问题
- 有一次遇到CPU跑20000步min还遇到这个问题改成100000步就好了